Research
The cell is the fundamental unit of biology, and long term changes involve a coordinated response which is ultimately determined by the genome. Today it is possible to obtain high-throughput, global, quantitative measurements of the genome, the transcriptome, the epigenome, and the proteome from individual cells. Analysis and integration of these large, complex, noisy datasets pose many challenges. As a computational genomics group, our goal is to develop machine learning methods that will provide new insights both for basic biology as well as for understanding how aberrant gene regulation plays a role in cancer, neurodegeneration and other diseases. We seek to develop scalable, robust, rigorous computational tools for tackling important biological problems. We are particularly interested in areas involving single-cell analysis, including multi-omics assay, spatial methods, and datasets from population studies.
Methods Development
We are interested in developing novel computational methods for analyzing high throughput sequencing data. In particular, there has been a focus on single-cell RNAseq, but we are also interested in other types of sequencing data.
Quantitative metrics for analyzing cellular architecture
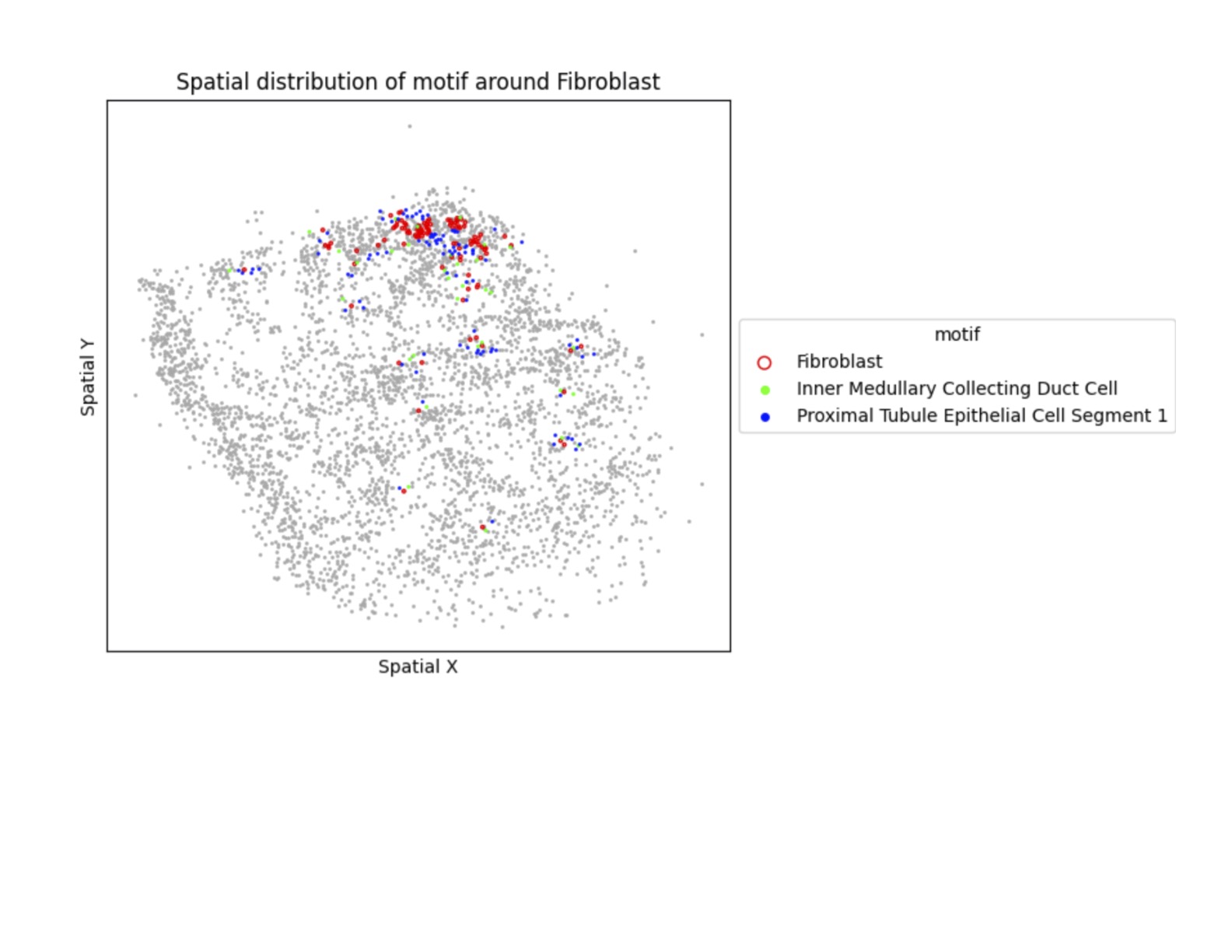
The latest generation of spatial transcriptomics methods make it possible to profile cells in their tissue context. The high-resolution spatial information allows us to investigate how cells are organized in the tissue. We are using ideas from spatial statistics to develop new approaches for understanding the organizational principles of cellular architecture. These methods make it possible to quantify differences in tissue structure, and will be helpful for understanding the impact of various disease conditions.
Batch correction and data integration
In collaboration with the Korsunsky and Raychaudhuri labs we are developing the next version of the popular Harmony package for merging single cell datasets to eliminate technical artefacts. The new version is orders of magnitudes faster and more memory efficient, allowing for the integration of tens of millions of cells from tens of thousands of batches. In addition, the upgraded method features automatic tuning of hyper parameters to avoid overcorrection.
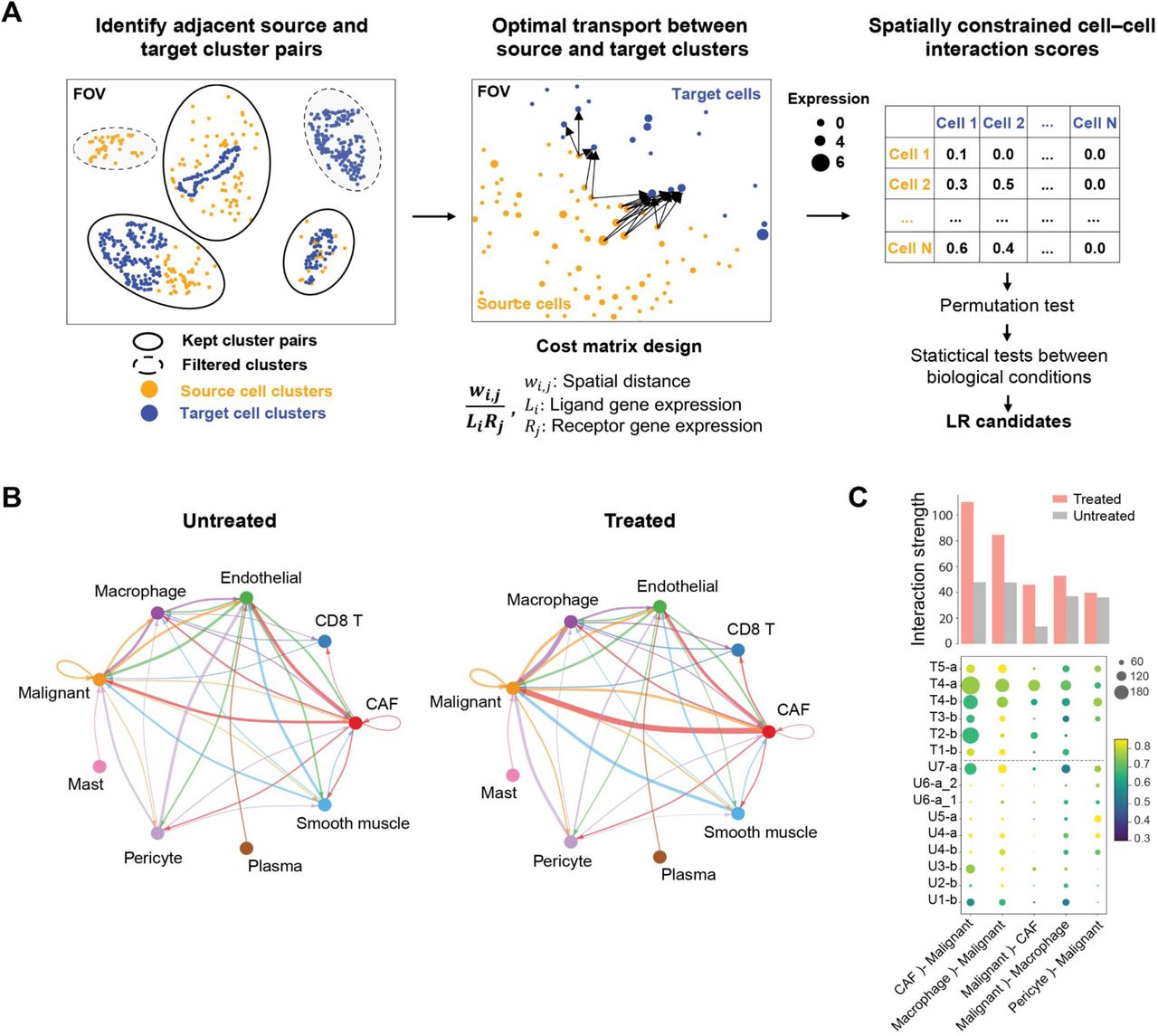
Inferring cell-cell communication from high-resolution spatial transcriptomics data
We are developing Scotia, a method for inferring cell-cell interactions via ligand-receptor communication. Scotia can leverage high-resolution spatial transcriptomics data to evaluate both the distance between cells as well as their similarity in terms of gene expression. As such, it is possible to exclude interactions that are implausible due to cells being located far apart. Predictions from Scotia for pancreatic adenocarcinoma have been experimentally validated, suggesting that it can identify relevant interactions.
Early detection of cancer using cell-free RNA
Liquid biopsies have emerged as a cost-effective and minimally invasive method for detecting cancer at an early stage. Through a simple blood draw, it is possible to detect molecules that have been shed from a tumor. To date, most work has focused on cell free DNA, but it is also possible to detect RNA molecules from the tumor. We are developing computational methods for detecting such signals from next generation sequencing data, and an important advantage of RNA over DNA is that it can also provide useful information about the malignant transcriptome.
Collaborative Projects
We work with several experimental groups, and most of our collaborations are in the areas of neuroscience, immunology and cancer.
Identification of drivers of treatment resistance in pancreatic cancer
In collaboration with the Hwang lab we are working on multiple projects related to pancreatic cancer, with a focus on understanding what are the drivers of therapeutic resistance. This is a common problem with this malignancy, and one of the main reasons behind the high mortality rates. We are analyzing high-resolution, high-plex spatial transcriptomics and spatial proteomics data to characterize the microenvironment, to understand how it varies across different malignant subtypes.
Determinants of influenza vaccine protection
We are working with the Marasco lab to investigate what are the molecular mechanisms underlying a strong immune protection following vaccination against the seasonal flu. This is particularly important for the elderly population where protection rates are the lower and mortality is the highest. By profiling PBMCs via single cell RNAseq following vaccinations, we aim to identify cell types and genes that are associated with a favorable response.
Perianal fistulizing disease
We are working with the Salas lab at IDIBAPS in Barcelona to profile patients with this subtype of inflammatory bowel disease. It is our hope that single-cell profiling will make it possible to identify biomarkers and therapeutic targets.
CAR T-cells for solid tumors
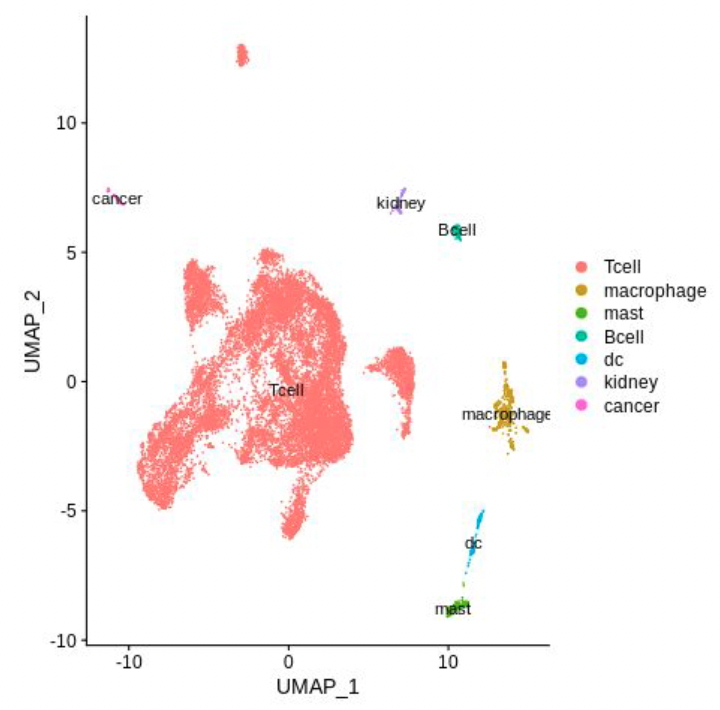
Together with the Marasco lab at the Dana-Farber Cancer Institute we are trying to understand the mechanisms involved in a novel design which is aimed at overcoming the suppresive tumor microenvironment. By profiling tumors using single cell RNAseq and spatial transcriptomics we can identify the cellular and molecular features that distinguish poor and good responders.